I høst sa Norge ja til å betale for det som kalles verdens dyreste medisin, genterapien Zolgensma. Men hva er egentlig genterapi, og hvordan fungerer det?
Av: Stine Hufthammer Indrelid og Eirik Joakim Tranvåg
Genterapi er en helt ny måte å behandle sykdom på og gir nytt håp om behandling for en rekke tilstander. Med genterapi kan man blant annet erstatte et gen som ikke fungerer eller skru av et gen som gir sykdom. To nye, revolusjonerende genterapier har nylig kommet på listen over behandlinger som det norske helsevesenet vil betale for; Zolgensma (spinal muskelatrofi, SMA) og Luxturna (arvelig synstap). Vi skal se nærmere på hvordan disse to genterapiene fungerer.
Det er viktig at behandling begynner tidlig.
Illustrasjonsfoto: iStock
Fjerne årsak til sykdom
I genene våre ligger oppskriften på proteiner, viktige byggesteiner som utfører de mange funksjonene som må til for at cellene og kroppen vår skal fungere. Noen ganger oppstår det feil i genene, og det kan gjøre at viktige proteiner enten ikke blir produsert, eller blir feildannet. Dette kan føre til sykdom. Slike genfeil kan være nedarvet, eller de kan oppstå spontant i noen av kroppens celler i løpet av livet. Konvensjonelle legemidler kan noen ganger erstatte et protein som mangler eller som er feilprodusert, men for en del sykdommer har slik behandling vist seg vanskelig og lite effektivt.
Gener med feil
Pasienter med muskelsykdommen SMA har en genfeil i det som heter SMN1-genet, noe som gjør at de motoriske nervecellene i ryggmargen ikke produserer nok SMN-protein. Dette fører til at cellene dør og musklene gradvis svekkes. Zolgensma motvirker dette ved å sette inn DNA med oppskriften for SMN1-genet i cellene, slik at cellene kan produsere proteinet som mangler. Luxturna er en genterapi mot arvelig synstap som skyldes feil i RPE65-genet. Denne genfeilen gjør at synet gradvis svekkes. Behandlingen tilfører DNA med oppskriften for en fungerende versjon av genet inn i synsceller på baksiden av øyet, under netthinnen. Dermed kan cellene i netthinnen selv produsere proteinet som mangler. Det vil bremse synstapet, og for mange også forbedre synet. For begge behandlingene, og andre genterapier, er det viktig at behandling settes i gang tidlig i sykdomsforløpet. For Zolgensma er det i hovedsak barn under seks måneder som er aktuell for behandling, og disse vil få diagnosen tidlig ettersom SMA nylig ble en del av nyfødtscreeningen i Norge. Arvelig synstap vil også som regel oppdages tidlig, gjennom synsundersøkelser på helsestasjon. Ofte ser disse barna godt de første årene og Luxturna vil først bli aktuelt når synet svekkes i rundt 10-års alderen. Hvor mye det norske helsevesenet skal betale for disse genterapiene er ukjent, men prisen før rabatt for Zolgensma er rundt 27 millioner kroner for én behandling. Les mer om godkjenning og vurdering i denne artikkelen.
Avanserte terapier:
Genterapi: overføring av nytt genetisk materiale til mennesker for å reparere eller erstatte et defekt gen.
Celleterapi: celler som blir gitt til mennesker for å behandle sykdom.
Vevsterapi: celler eller vev som blir gitt til mennesker for å reparere eller erstatte vev eller organer.
Avanserte terapier
Avanserte terapier er en samlebetegnelse på nye typer behandlinger som blant annet inkluderer genterapier. I genterapier som Zolgensma pakkes det genetiske materialet med «erstatningsgenet» inn i et ufarlig virus (også kalt virusvektor) som så gis direkte inn i pasienten (in vivo). Det genetiske materialet fraktes inn i cellekjernene av cellenes egne transportsystemer, og dermed blir den riktige oppskriften til SMN-proteinet igjen tilgjengelig for cellene. For andre typer genterapi, som immunterapien CAR-T mot kreft, kombineres genterapi og celleterapi. Først tar man immunceller ut av kroppen (ex vivo). Deretter bruker man en virusvektor til å sette genetisk materiale inn i pasientens immunceller slik at de kjenner igjen kreftcellene. Til slutt tilbakeføres disse modifiserte immuncellene til pasienten. Pasientens immunforsvar blir da genetisk programmert til å bekjempe kreftcellene i sin egen kropp.
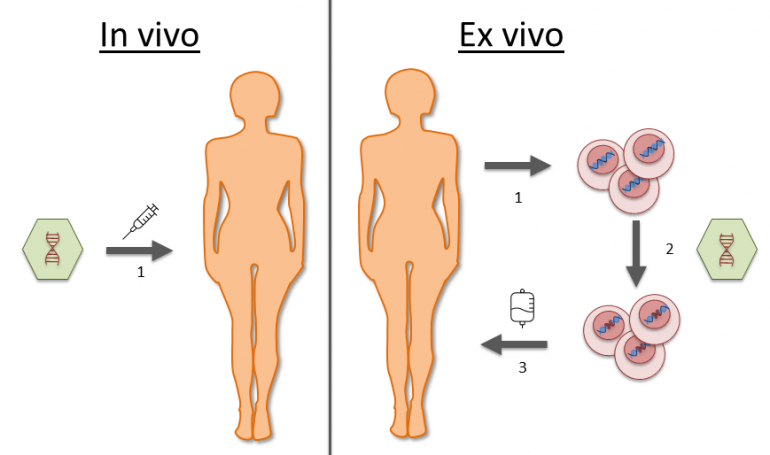
Ex vivo genterapi: 1. Celler hentes ut fra pasienten. 2. En virusvektor med genetisk materiale
modifiserer cellene. 3. De genmodifiserte cellene gis tilbake til pasienten. Figur: Eirik Joakim Tranvåg
Optimisme og tilbakeslag
Å bruke virusvektor til å transportere det genetiske materialet inn i cellene, var også sentral i utviklingen av de første genterapiene som ble testet på mennesker i 1990- årene. Genterapifeltet ble imidlertid rammet av flere alvorlige tilbakeslag nesten før det var kommet i gang. Noen av behandlingene som ble testet ut viste seg å ha dårlig effekt, og flere pasienter utviklet alvorlige bivirkninger etter å ha blitt behandlet med datidens genterapi. En 18-åring som deltok i en klinisk studie døde da han fikk en kraftig immunrespons mot virusvektoren som var brukt til å føre genterapien inn i cellene hans. Men i tiden som har gått siden genterapiens begynnelse på 1990-tallet har man fått mye ny kunnskap både om menneskets genom og den genetiske bakgrunnen for sykdom. Samtidige har det blitt jobbet med å finne tryggere og mer effektive metoder for å få genterapien inn i cellene. Som et resultat av denne prosessen har man fått disse nye genterapiene som nå er godkjent, og en lang rekke andre genterapier er under utprøving og utvikling.
Med genredigering er det teknisk mulig å korrigere genfeil i befruktet egg som skal danne nye individer.
Genredigeringsterapi
I 2012 beskrev de to forskerne Emanuelle Charpentier og Jennifer Doudna en ny metode for å gjøre endringer i genene der de er; genredigering med Crispr. Metoden blir omtalt som en gensaks som ved hjelp av et protein, Cas-9, kan brukes til å klippe og redigere på et ønsket sted i genomet. Dette kan løse utfordringene med at genterapier som bruker virus for å transportere inn et «erstatningsgen» setter dette inn på et tilfeldig sted i genomet, eller at genet blir liggende utenfor genomet og derfor kun gir midlertidig effekt. Slik genredigeringsterapi gir også flere fordeler: Der man ved førstegenerasjons genterapier kun kan gjøre en type modifikasjon – nemlig å sette inn nytt genetisk materiale i celler, gir genredigering flere muligheter. Man kan for eksempel «skru av» en genvariant som gir sykdom eller gjøre mindre korreksjoner i en gensekvens i cellene. Når en bruker genredigering på denne måten endrer man genomet i cellen permanent. I august i år kom resultatene av en studie der nettopp genredigering ble brukt direkte i pasienter. Pasientene hadde en livstruende leversykdom hvor en genfeil fører til overproduksjon av et protein som heter transtyretin, som hoper seg opp i nerveceller og hjertevev. Forskerne programmerte Crispr-Cas9-proteinet til å kjenne igjen og endre genfeilen i levercellene. Genredigeringsterapien ble gitt intravenøst til seks pasienter, i en engansbehandling, og resultatene av studien viste at terapien ble tatt opp av levercellene. Alle seks pasienter hadde tydelig lavere nivåer av det feildannede proteinet etter behandling og ingen av pasientene fikk alvorlige bivirkninger. Forskerne bak studien understreker imidlertid at pasientene skal følges opp over lengre tid.
Utrydde arvelig sykdom?
Men hva med å bruke genterapi for å ta vekk genfeil også i kjønnsceller, slik at en alvorlig, arvelig sykdom ikke lenger vil gå i arv? Med genredigering er det teknisk mulig å korrigere genfeil i befruktet egg som skal danne nye individer. Samtidig reiser en slik endring av arvelige genetiske egenskaper i mennesker mange nye og vanskelige etiske problemstillinger. Hvordan vet vi at slik behandling er trygg? Når kan vi vite det? Hvordan kan et ikke-eksisterende individ samtykke til behandling? Nok en gang er det tydelig at de mulighetene som ny medisinsk bioteknologi gir oss også fører til nye etiske utfordringer. Du kan lese mer om den internasjonale debatten rundt dette i denne GENialt-artikkelen.


